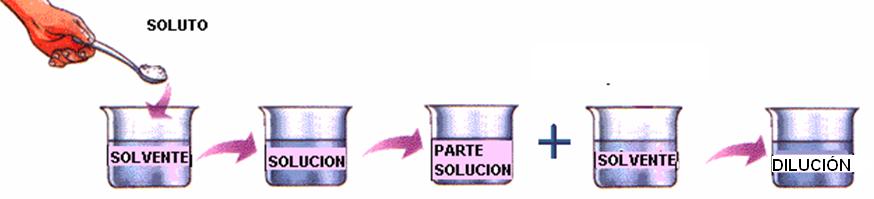
DILUCIONES
La dilución es el procedimiento que se sigue para preparar una disolución menos concentrada a partir de una más concentrada.
Definición general de disolución:Una dilución es una mezcla homogénea, uniforme y estable, formada por dos o más sustancias denominadas componentes. La sustancia presente en mayor cantidad suele recibir el nombre de solvente, y a la de menor cantidad se le llama soluto y es la sustancia disuelta. El soluto puede ser un gas, un líquido o un sólido, y el disolvente puede ser también un gas, un líquido o un sólido.
Observadas a través del microscopio, las diluciones aparecen homogéneas y el soluto no puede separarse por filtración.
Es una mezcla ya que las cantidades de los componentes no son fijas y también se denomina mezcla por que no hay reacción química en la unión de componentes.
Se denomina Homogénea porque:
Es uniforme ante la observación visual directa o con microscopio, y no apreciamos la existencia de varias partes o fases.
Las partículas de los componentes son de tamaño molecular y están distribuidas sin ningún orden.
Es una mezcla ya que las cantidades de los componentes no son fijas y también se denomina mezcla por que no hay reacción química en la unión de componentes.
Se denomina Homogénea porque:
Es uniforme ante la observación visual directa o con microscopio, y no apreciamos la existencia de varias partes o fases.
Las partículas de los componentes son de tamaño molecular y están distribuidas sin ningún orden.
Se denomina Uniforme puesto que en todas sus partes tiene una misma composición con las mismas propiedades, cojamos la porción de mezcla que cojamos en cada una de ellas siempre encontraremos el mismo contenido en cuanto a sus componentes. Además las partículas se hallan distribuidas de forma ordenada, y no al azar.Se denomina Estable por mantenerse en su composición inicial sin cambiar en cuanto a los componentes químicos que la forman.
Soluto y disolvente:
Soluto y disolvente:
Soluto: Es el componente que cambia de fase cuando se produce la disolución; también denominado cuerpo disperso.
Solvente: Es el componente que disuelve, teniendo la propiedad de disolver ciertas sustancias.

1.1 NORMAS PARA LA ELECCIÓN DE SOLUTO Y SOLVENTE:
La sustancia presente en mayor cantidad suele recibir el nombre de disolvente, y a la de menor cantidad se le llama soluto y es la sustancia disuelta. El soluto puede ser un gas, un líquido o un sólido, y el disolvente puede ser también un gas, un líquido o un sólido. El agua con gas es un ejemplo de un gas (dióxido de carbono) disuelto en un líquido (agua). Las mezclas de gases, como ocurre en la atmósfera, son disoluciones.
En las disoluciones entre un sólido y un líquido es fácil identificar el soluto y el disolvente; pero si se trata de dos o más líquidos o gases, la distinción entre soluto y disolvente es arbitraria.
1.2 SOLUBILIDAD
La solubilidad de un compuesto en un solvente concreto y a una temperatura y presión dadas se define como la cantidad máxima de ese compuesto que puede ser disuelta en la dilución. En la mayoría de las sustancias, la solubilidad aumenta al aumentar la temperatura del disolvente. En general, la mayor solubilidad se da en disoluciones cuyas moléculas tienen una estructura similar a las del solvente.
La sustancia presente en mayor cantidad suele recibir el nombre de disolvente, y a la de menor cantidad se le llama soluto y es la sustancia disuelta. El soluto puede ser un gas, un líquido o un sólido, y el disolvente puede ser también un gas, un líquido o un sólido. El agua con gas es un ejemplo de un gas (dióxido de carbono) disuelto en un líquido (agua). Las mezclas de gases, como ocurre en la atmósfera, son disoluciones.
En las disoluciones entre un sólido y un líquido es fácil identificar el soluto y el disolvente; pero si se trata de dos o más líquidos o gases, la distinción entre soluto y disolvente es arbitraria.
1.2 SOLUBILIDAD
La solubilidad de un compuesto en un solvente concreto y a una temperatura y presión dadas se define como la cantidad máxima de ese compuesto que puede ser disuelta en la dilución. En la mayoría de las sustancias, la solubilidad aumenta al aumentar la temperatura del disolvente. En general, la mayor solubilidad se da en disoluciones cuyas moléculas tienen una estructura similar a las del solvente.
1.3 PROPIEDADES FÍSICAS DE LAS DILUCIONES
Cuando se añade un soluto a un disolvente, se alteran algunas propiedades físicas del disolvente. Al aumentar la cantidad del soluto, sube el punto de ebullición y desciende el punto de solidificación.
Otra propiedad destacable de una disolución es su capacidad para ejercer una presión osmótica. Si separamos dos diluciones de concentraciones diferentes por una membrana semipermeable (una membrana que permite el paso de las moléculas del disolvente, pero impide el paso de las del soluto), las moléculas del disolvente pasarán de la disolución menos concentrada a la disolución de mayor concentración, haciendo a esta última más diluida.
Cuando se añade un soluto a un disolvente, se alteran algunas propiedades físicas del disolvente. Al aumentar la cantidad del soluto, sube el punto de ebullición y desciende el punto de solidificación.
Otra propiedad destacable de una disolución es su capacidad para ejercer una presión osmótica. Si separamos dos diluciones de concentraciones diferentes por una membrana semipermeable (una membrana que permite el paso de las moléculas del disolvente, pero impide el paso de las del soluto), las moléculas del disolvente pasarán de la disolución menos concentrada a la disolución de mayor concentración, haciendo a esta última más diluida.

1.4 CONCENTRACIÓN DE UNA DISOLUCIÓN
Existen distintas formas de expresar la concentración de una dilución, pero las dos más utilizadas son: gramos por litro (g/l) y molaridad (M). Los gramos por litro indican la masa de soluto, expresada en gramos, contenida en un determinado volumen de disolución, expresado en litros. Así, una disolución de cloruro de sodio con una concentración de 40 g/l contiene 40 g de cloruro de sodio en un litro de disolución.
Existen distintas formas de expresar la concentración de una dilución, pero las dos más utilizadas son: gramos por litro (g/l) y molaridad (M). Los gramos por litro indican la masa de soluto, expresada en gramos, contenida en un determinado volumen de disolución, expresado en litros. Así, una disolución de cloruro de sodio con una concentración de 40 g/l contiene 40 g de cloruro de sodio en un litro de disolución.
La molaridad se define como la cantidad de sustancia de soluto, expresada en moles, contenida en un cierto volumen de disolución, expresado en litros, es decir: M = n/V. El número de moles de soluto equivale al cociente entre la masa de soluto y la masa de un mol (masa molar) de soluto

Dos disoluciones de KMno4 a diferente concentración.
2. DILUCIONES SERIADAS
Hay muchas situaciones en que las cantidades de sustancia necesarias para ver un efecto son extremadamente pequeñas (por ejemplo un fármaco para tratar una enfermedad, una hormona para estudiar su efecto en un animal de experimentación, etc) y difícilmente se pueden pesar o medir en esas proporciones tan pequeñas. En esas situaciones es necesario recurrir a la preparación de una solución de alta concentración (o solución de stock o solución madre) y hacer diluciones seriadas a partir de ésta.
2.1 ¿CÓMO SE PREPARAN LAS DILUCIONES SERIADAS?
2.1 ¿CÓMO SE PREPARAN LAS DILUCIONES SERIADAS?
En general se parte de una solución concentrada y se preparan series de diluciones al décimo (1:10) o al medio (1:2). De esta manera se obtiene una serie de soluciones relacionadas por ejemplo por un factor de dilución 10 es decir 1/10; 1/100; 1/1000 y así sucesivamente. O la otra serie es 1/2; 1/4; 1/8; 1/16; 1/32 etc. Por ejemplo: si partimos de una solución de 50mg/ml de una sustancia (Solución A)
a. Dilución 1/10: 1ml de la solución A + 9 ml de agua= una solución de 5 mg/ml (dilución 1:10)
b. Dilución 1/2: 5 ml de la solución A + 5 ml de agua= una solución de 25 mg /ml (dilución 1:2)











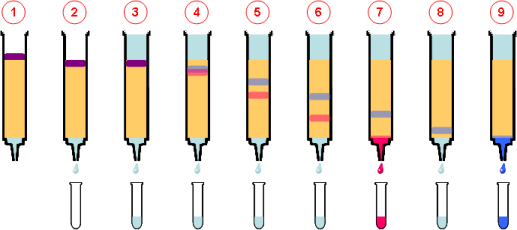
 Las moléculas amfotéricas, como los aminoácidos, se separan en un medio en el que existe una diferencia de potencial y un gradiente de pH . La región del ánodo es Ácida y la del cátodo es alcalina. Entre ambos se establece un gradiente de pH tal que las moléculas que se han de separar tenga su punto isoeléctrico dentro del rango. Las sustancias que inicialmente se encuentran en regiones de pH inferior a su punto isoeléctrico estarán cargadas positivamente y migraran hacia el cátodo, mientras aquellas que se encuentran en medios con pH más bajos que su punto isoeléctrico tendrán carga negativa y migraran hacia el ánodo. La migración les conducirá a una región dónde el pH coincidirá con su punto isoléctrico, tendrán una carga neta nula y se detendrán. De esta forma las moléculas amfotéricas se sitúan en estrechas bandas donde coincide su punto isoeléctrico con el pH.
Las moléculas amfotéricas, como los aminoácidos, se separan en un medio en el que existe una diferencia de potencial y un gradiente de pH . La región del ánodo es Ácida y la del cátodo es alcalina. Entre ambos se establece un gradiente de pH tal que las moléculas que se han de separar tenga su punto isoeléctrico dentro del rango. Las sustancias que inicialmente se encuentran en regiones de pH inferior a su punto isoeléctrico estarán cargadas positivamente y migraran hacia el cátodo, mientras aquellas que se encuentran en medios con pH más bajos que su punto isoeléctrico tendrán carga negativa y migraran hacia el ánodo. La migración les conducirá a una región dónde el pH coincidirá con su punto isoléctrico, tendrán una carga neta nula y se detendrán. De esta forma las moléculas amfotéricas se sitúan en estrechas bandas donde coincide su punto isoeléctrico con el pH.
 La electroforesis es una técnica muy sensible y puede ser afectada por muchos errores experimentales, como la temperatura durante la polimerización y la corrida del gel, velocidad de la polimerización, niveles de catalizador, pureza del reactivo, tiempo de corrida y preparación de las muestras.
La electroforesis es una técnica muy sensible y puede ser afectada por muchos errores experimentales, como la temperatura durante la polimerización y la corrida del gel, velocidad de la polimerización, niveles de catalizador, pureza del reactivo, tiempo de corrida y preparación de las muestras.